
Tout comme Alzheimer, la maladie de Charcot est une maladie neurodégénérative grave, rapidement évolutive et particulièrement difficile à vivre au quotidien.
Dans cet article, nous vous présentons les origines de cette maladie rare, ses différents symptômes, les examens qui permettent de la diagnostiquer et les traitements utilisés pour apaiser les troubles qui y sont associés.
Quelques éléments de définition
Aussi connue sous le nom de sclérose latérale amyotrophique (SLA), la maladie de Charcot est une maladie rare et neurodégénérative particulièrement invalidante au quotidien. Elle survient généralement à l’âge adulte entre 50 et 70 ans environ.
Cette maladie se caractérise par une paralysie croissante des muscles qui sont impliqués dans la motricité volontaire. D’après les scientifiques, cette paralysie est due à la disparition de neurones moteurs, appelés aussi « motoneurones », qui sont des cellules nerveuses spécialisées dans le contrôle des mouvements volontaires. C’est cette disparition des neurones qui entraîne une atrophie des muscles jusqu’à une paralysie complète des membres.
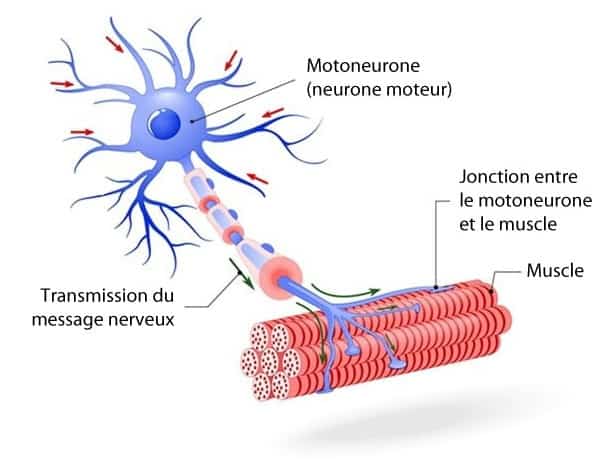
Il faut savoir que cette maladie touche également la phonation (qui permet la production du langage et des sons) ainsi que la déglutition (qui permet de se nourrir et de s’hydrater).
Généralement, elle entraîne un pronostic vital très sévère puisque la personne qui en souffre décède dans les 3 à 5 années suivant le diagnostic. Bien souvent, c’est l’atteinte des muscles respiratoires (en particulier du diaphragme) qui en est la cause.
Bien plus rarement, il existe aussi certaines formes plus bénignes de la maladie qui peuvent rester stables pendant une trentaine d’années puis se dégrader plus tard.
💡 Bon à savoir
Si vous cherchez une solution pour permettre à un proche atteint de la maladie de Charcot de garder facilement contact avec sa famille, nous vous recommandons LiNote. Sans jamais devoir y toucher, cet appareil lui permettra de recevoir des photos, vidéos, messages et de discuter en visio. Il pourra aussi appeler quelqu’un sans avoir à composer son numéro de téléphone grâce aux touches raccourci.
Qu’est-ce que la motricité volontaire ?
La motricité volontaire peut se définir comme l’activité nerveuse qui permet l’exécution de mouvements contrôlés par le cerveau. Pour résumer, il s’agit des mouvements que nous effectuons volontairement. Par exemple, le fait de saisir une tasse avec sa main sollicite la motricité volontaire.
À contrario, certains muscles de notre corps ne peuvent pas être commandés volontairement. C’est par exemple le cas du muscle cardiaque qui fonctionne de manière indépendante et autonome, sans que nous ayons besoin d’y penser.
Dans la maladie de Charcot, deux types de neurones moteurs impliqués dans la motricité sont touchés :
- les neurones moteurs dits centraux, qui sont situés dans le cerveau. Ces neurones acheminent le message provenant des neurones situés dans le cerveau jusqu’aux neurones situés dans la moelle épinière.
- les neurones moteurs dits périphériques, qui sont localisés dans la moelle épinière et le tronc cérébral. Ces neurones permettent d’assurer la correspondance entre les neurones moteurs centraux et les muscles volontaires.
💡 Le saviez-vous ?
Le nom Charcot a été donné à cette maladie suite aux découvertes du neurologue français Jean-Martin Charcot, qui l’a décrite au cours des années 1860.
Dans le milieu médical, on parle aussi de « sclérose latérale amyotrophique » pour les raisons suivantes :
– Le terme « sclérose » fait référence à l’apparition d’un tissu fibreux et sclérosé qui remplace les motoneurones disparus. Ce tissu peut être observé à l’aide d’un microscope.
– Le terme « latérale » fait référence au trajet emprunté par la maladie lors de la dégénérescence des cellules nerveuses, autrement dit celles du cerveau (les neurones moteurs centraux) et de la moelle épinière (les neurones moteurs périphériques).
– Le terme « amyotrophique » fait référence aux conséquences de la maladie qui entraîne une perte en trophicité (autrement dit, en nutrition et en croissance) qui conduit ensuite à une maigreur des muscles et à leur paralysie progressive.
Enfin, aux États-Unis et au Québec, on l’appelle aussi la « maladie de Lou Gehrig » en hommage au célèbre joueur de baseball des Yankees, qui en est décédé en 1941.
La maladie de Charcot en chiffres
En France, on compte environ 8 000 personnes atteintes de la maladie de Charcot. Elle apparaît généralement entre 50 et 70 ans, et le plus souvent vers l’âge de 60-65 ans. Elle peut également apparaître plus précocement si elle est d’origine familiale. Bien qu’elle touche les deux sexes, sa prévalence est faiblement masculine, c’est-à-dire qu’elle touche un peu plus d’hommes que de femmes.
Quels sont les différents symptômes de la maladie de Charcot ?
De manière générale, les principaux symptômes de la maladie de Charcot se caractérisent par des troubles moteurs associés à :
- une atrophie musculaire avec une perte de la force ;
- une lenteur des mouvements ;
- et une certaine spasticité (raideur musculaire).
Toutefois, il arrive que d’autres symptômes s’ajoutent comme :
- des oedèmes (présence de liquide en dehors des vaisseaux sanguins) ou des troubles vasomoteurs (troubles de la circulation sanguine liés à un relâchement des vaisseaux sanguins (rougeurs) ou à leur compression (pâleur)) ;
- des troubles du sommeil ;
- des troubles de l’alimentation (avec amaigrissement et constipation).v
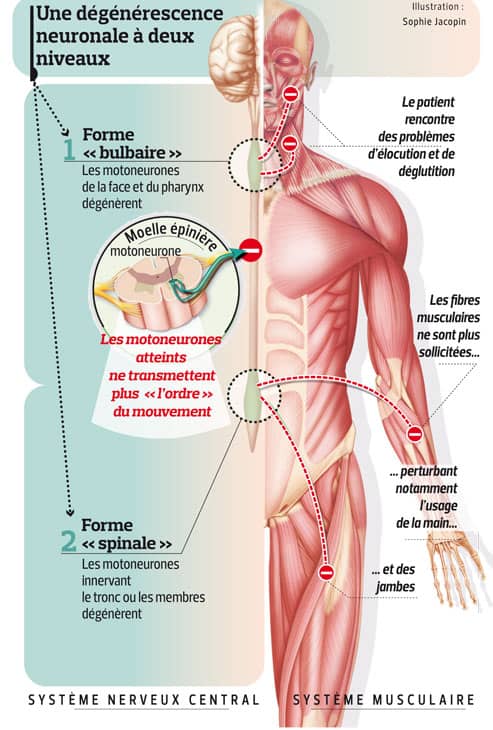
Il va sans dire que tous ces symptômes peuvent avoir des conséquences fonctionnelles très importantes chez la personne qui en souffre. Généralement, les symptômes de la maladie démarrent au niveau d’une région corporelle particulière (comme la main, le pied ou encore la bouche par exemple) puis s’étendent à l’ensemble du corps. Selon la zone du corps initialement atteinte, on distingue deux formes de la maladie : la forme spinale et la forme bulbaire.
La forme spinale
La maladie commence par atteindre les motoneurones situés dans la moelle épinière cervicale, dorsale et lombaire. Ainsi, elle entraîne :
- des troubles de la motricité :
– des membres supérieurs (bras, avants-bras, poignets, mains) ;
– des membres inférieurs (hanches, cuisses, jambes, pieds) ;
– et du diaphragme, pouvant entraîner d’importantes conséquences respiratoires ; - des troubles de l’équilibre ;
- des trouble de la coordination ;
- des douleurs (liées à des crampes, contractures, raideurs) dans les membres et leurs articulations ;
- de l’anxiété, voire de la dépression.
Ces troubles finissent par atteindre l’ensemble des membres, mais souvent de façon asymétrique. Autrement dit, un côté est généralement plus touché que l’autre.
💡 Bon à savoir
Cette forme de la maladie est la plus fréquente (environ 2/3 des cas). Elle affecte plus souvent les hommes et démarre généralement autour de 55-60 ans.
La forme bulbaire
La maladie commence par atteindre les motoneurones situés dans le tronc cérébral. Ainsi, les premiers symptômes de la maladie sont :
- des troubles de la parole composés de difficultés à prononcer certains mots, à articuler avec parfois des changements de voix (voix faible ou rauque) ;
- des troubles de la déglutition qui entraînent des difficultés à mâcher les aliments, à bouger la langue et à avaler, ce qui rend difficile la capacité à s’alimenter et à s’hydrater ;
- une hypersalivation ou au contraire une sécheresse de la bouche ;
- et dans certains cas, une alternance de moments de rires ou de pleurs involontaires.
💡 Bon à savoir
Cette forme de la maladie est la plus rare. Elle apparaît plus fréquemment chez les femmes et démarre autour de 60-65 ans.
L’évolution des symptômes
La vitesse à laquelle se diffuse ces symptômes varie d’un individu à un autre, ce qui rend l’évolution de la maladie difficilement prévisible.
Cependant, la maladie évolue toujours vers une forme complète, associant progressivement les symptômes de la forme spinale à ceux de la forme bulbaire, ou inversement.
Il semble également important de préciser que quelle que soit la forme de la maladie, les fonctions sensorielles, intellectuelles, sexuelles et urinaires restent préservées.
Un suivi régulier de la maladie par une équipe de professionnels de la santé est véritablement indispensable. Ce suivi permet notamment de porter une attention toute particulière à :
- La gêne respiratoire car si les muscles respiratoires sont touchés, les risques d’infections et d’insuffisance respiratoire sont importants.
- L’évolution de la paralysie des membres car la maladie entraîne d’importants risques de phlébite, d’embolie pulmonaire, de douleurs, de raideurs et autres blocages articulaires.
- L’évolution des troubles de la déglutition car ils peuvent malencontreusement conduire à une infection pulmonaire ou faire survenir une dénutrition (insuffisance d’apports nutritionnels).
Comment se déclenche la maladie de Charcot ?
Une origine familiale
Dans 10% des cas, l’origine de la maladie est héréditaire avec une transmission autosomique dominante, c’est-à-dire qu’elle peut se transmettre de génération en génération. Dans cette situation, elle se déclare assez tôt, en moyenne avant l’âge de 50 ans. Elle peut notamment être liée à une ou plusieurs mutations sur les gènes suivants :
- le gène de la superoxide dismutase 1 ou SOD1 (cette mutation concerne près de 15% des formes familiales) situé sur le chromosome 21 ;
- le gène C9ORF72 situé sur le chromosome 9 ;
- le gène TARDBP situé sur le chromosome 1 ;
- le gène FUS/TLS situé sur le chromosome 16 ;
- le gène TBK1 situé sur le chromosome 12.
Il faut savoir que bien souvent, la forme familiale de cette maladie est liée à l’altération de plusieurs gènes à la fois (on parle alors de « forme multigénique »). Il n’est donc pas toujours évident d’identifier l’origine génétique des formes familiales de la maladie.
Une origine sporadique
Mais dans la grande majorité des cas, la maladie de Charcot est de forme sporadique, c’est-à-dire qu’elle apparaît sans aucune raison héréditaire et qu’elle n’est pas transmissible.
Toutefois, les facteurs génétiques présentés ci-dessus peuvent une fois encore constituer des facteurs de risques, c’est-à-dire favoriser l’apparition de la maladie de Charcot.
Par ailleurs, d’autres facteurs, cette fois-ci environnementaux peuvent également contribuer au développement de la maladie. C’est pour ces raisons que l’on dit de cette maladie qu’elle est, dans la plupart des cas, multifactorielle.
Quels sont les facteurs environnementaux en jeu dans le déclenchement de la maladie de Charcot ?
Même si aucun facteur environnemental n’a été distinctement mis en évidence, voici ceux qui sont à ce jour susceptibles de contribuer au déclenchement de la maladie de Charcot :
- le tabac ;
- l’exposition à certains produits agricoles chimiques (comme les pesticides), métaux lourds (comme le plomb et le mercure) et algues (comme la cyanotoxine BMAA) ;
- une activité physique très intense et importante ;
- des traumatismes répétés.
D’après les chercheurs, ces facteurs environnementaux pourraient contribuer à faire dysfonctionner les motoneurones, ce qui entraînerait progressivement l’apparition de la maladie.
Comment diagnostiquer la maladie de Charcot ?
Tout d’abord, il faut savoir que cette maladie est particulièrement difficile à diagnostiquer en raison de son absence de marqueurs biologiques associés (comme une anomalie sanguine, radiologique ou neurophysiologique particulière qui permettraient de repérer immédiatement la maladie, par exemple).
Le diagnostic s’effectue donc à partir d’un certain nombre d’examens cliniques et neurologiques.
La détection de signes cliniques
Un neurologue commence par repérer la présence éventuelle de signes cliniques d’atteinte :
- des motoneurones centraux
raideur des muscles, amplification des réflexes, détection éventuelle d’un signe de Babinski ;
- des motoneurones périphériques
diminution du volume des muscles (amyotrophie), crampes, disparition des réflexes, contractions involontaires des muscles (fasciculations).
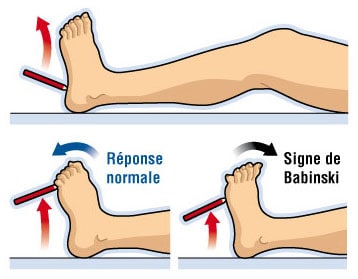
Qu’est-ce qu’un signe de Babinski ?
Le signe de Babinski (ou réflexe cutané plantaire) correspond à un signe clinique qui permet de détecter la présence d’une lésion au sein du système nerveux central. Il consiste à stimuler le pied à l’aide d’un objet fin (comme un stylo) avec un mouvement allant de la plante du pied vers les orteils.
Lorsqu’un patient ne présente pas de problèmes neurologiques, son réflexe est de fléchir ses orteils, le gros orteil s’orientant alors vers la plante du pied. Lorsqu’un patient présente effectivement un problème neurologique, son réflexe est de tendre ses orteils, en particulier le gros orteil.
Un panel d’examens poussés
Par la suite, l’équipe médicale complète ce premier examen par :
- Un bilan biologique afin d’éliminer la présence de troubles immunitaires ou métaboliques et de relever le taux de créatine phosphokinase (CPK). Sa quantité anormalement élevée permet de mettre en évidence la présence d’une faiblesse musculaire importante. Ce bilan permet également de vérifier le dosage des neurofilaments (Nf). Un taux particulièrement élevé dans le sang ou dans le liquide céphalo-rachidien (LCR) témoigne de la destruction d’un certain nombre de motoneurones.
- Une IRM (Imagerie par Résonance Magnétique) médullaire (de la moelle épinière) et parfois cérébrale (du cerveau) pour permettre d’écarter les causes organiques.
- Un examen neurologique de référence, appelé l’« électromyogramme » (ou EMG) pour éliminer d’autres maladies potentielles et confirmer le diagnostic de la maladie de Charcot.
Comment se déroule un électromyogramme ?
Cet examen peut être réalisé auprès d’un neurologue exerçant dans un cabinet privé ou à l’hôpital (au sein d’un service de neurophysiologie). Ce dernier permet d’étudier l’activité électrique des nerfs et des muscles.
Dans le cas de la maladie de Charcot, l’électromyogramme peut révéler un bon fonctionnement des muscles mais un affaiblissement des nerfs dont les motoneurones.
L’examen dure entre 45 et 90 minutes. L’appareil utilisé pour réaliser cet examen neurologique se nomme un « electromyographe ».
Tandis que la personne qui passe cet examen s’allonge, le neurologue est chargé de disposer des électrodes semblables à de petits patchs sur la peau, à différents endroits. Ensuite, ces électrodes envoient de légères impulsions électriques (brèves et de faible intensité) dans le but d’analyser les capacités de transmission des nerfs. L’activité électrique est retransmise sur un appareil permettant une analyse sous forme de tracés.
Des examens complémentaires
Si ces examens ne suffisent pas à confirmer le diagnostic, d’autres examens spécifiques et complémentaires peuvent être prescrits comme :
- une ponction lombaire du liquide céphalo-rachidien (LCR) pour détecter la présence éventuelle d’une anomalie immunitaire, d’une infection ou encore d’une inflammation ;
- une biopsie musculaire pour identifier l’origine de l’atrophie musculaire, autrement dit, savoir si elle provient du muscle ou du système nerveux.
Quel traitement pour la maladie de Charcot ?
La plupart du temps, les personnes qui sont atteintes de la maladie de Charcot bénéficient d’une prise en charge pluridisciplinaire de leur santé. Ils peuvent notamment être soutenus par un(e) neurologue, un(e) ergothérapeute, un(e) kinésithérapeute, un(e) neuropsychologue, un(e) psychologue clinicien(ne), un(e) pneumologue, un(e) infirmier(e), etc.
Aujourd’hui, en France, il existe une vingtaine de centres de référence qui proposent une prise en charge complète et pluridisciplinaire de la maladie. Voici la liste des centres labellisés et spécialisés dans cette maladie.
Pour apaiser les symptômes
Bien qu’il n’existe pas de traitement qui permette de guérir complètement de la maladie de Charcot, certaines options thérapeutiques médicamenteuses et non médicamenteuses peuvent contribuer à apaiser les symptômes.
Les solutions non médicamenteuses
Parmi les solutions non médicamenteuses, on retrouve :
- des séances de rééducation ou de kinésithérapie
pour éduquer la personne afin qu’elle acquiert un meilleur équilibre, une plus grande souplesse de ses muscles, un maintien de son autonomie et qu’elle parvienne à mieux prévenir les chutes ;
- un suivi psychologique
pour soutenir et accompagner la personne dans les moments difficiles et l’aider à gérer ses émotions ;
- un suivi orthophonique
pour apaiser les troubles de la phonation, de la déglutition et aider la personne à maintenir la communication avec son entourage ;
- des consultations auprès d’un(e) diététicien(ne)
pour mettre en place un régime alimentaire adapté aux besoins nutritionnels de la personne atteinte de la maladie ;
- des soins dentaires
effectués par un dentiste qui connaît bien la maladie pour permettre à la personne de conserver une bonne hygiène dentaire et aspirer les résidus d’aliments ainsi que les excès de salive en cas d’hypersalivation ;
- des techniques de relaxation
pour l’aider à étirer et assouplir ses muscles, se relaxer et apprendre à se sentir apaisé malgré la maladie.
Les solutions médicamenteuses
Pour apaiser :
- les crampes et les douleurs musculaires :
certains médicaments décontractants comme les myorelaxants (baclofène, dantrolène) ou les antidouleurs (paracétamol, codéine voire morphine) peuvent s’avérer efficaces.
- l’anxiété et les symptômes dépressifs :
les antidépresseurs en complément d’un suivi psychothérapeutique peuvent être recommandés.
- les troubles du sommeil :
les benzodiazépines en compléments de séances de relaxation peuvent représenter une solution.
- la constipation :
des médicaments laxatifs tels que le sorbitol ou le lactulose peuvent faciliter le transit intestinal.
- l’hypersalivation :
certains médicaments comme la clomipramine, l’atropine ou encore l’ipratropium bromure peuvent aider à épaissir la salive ou à assécher la bouche. En ciblant directement les glandes salivaires, la radiothérapie peut aussi représenter une alternative pour réduire l’hypersalivation.
- les infections pulmonaires :
des antibiotiques peuvent parfois être prescrits en prévention si la personne a tendance à être victime de fausses routes alimentaires.
Pour améliorer la qualité de vie
Pour améliorer leur qualité de vie en fonction de l’évolution de leurs symptômes, les personnes atteintes de la maladie de Charcot peuvent avoir recours à des aides techniques et matérielles comme :
- un déambulateur qui permet d’offrir un premier appui rigide à la personne si elle présente des difficultés à marcher ;
- une orthèse qui permet de compenser et soutenir la fonction d’une articulation ou d’un muscle (ex : une gouttière, une attelle, un corset, etc.) ;
- un fauteuil roulant qui peut s’avérer difficile à accepter mais qui peut permettre à la personne d’économiser son énergie et d’être plus autonome au quotidien ;
- l’aménagement de son domicile (barres d’appui, monte-escaliers, lavabo suspendu, WC surélevé, etc.).
Pour améliorer l’espérance de vie
Bien que le pronostic de la maladie reste sévère, certaines techniques peuvent contribuer à améliorer l’espérance de vie des personnes atteintes de la maladie de Charcot. Parmi les principales, on retrouve :
- la prescription de riluzole, un médicament qui agit directement sur le système nerveux central et qui permet de réduire la destruction des motoneurones ;
- la prescription de vitamine E, reconnue pour ses vertus antioxydantes ;
- l’installation d’une gastrostomie qui correspond à un tuyau en plastique permettant de relier l’estomac à la paroi extérieure du ventre. Cet appareil permet d’introduire directement des aliments liquides dans l’estomac afin d’éviter les fausses routes alimentaires et réduire le risque d’infection respiratoire ;
- la ventilation non invasive (ou VNI), une assistance ventilatoire présentée sous forme de masque (nasal, narinaire, bucco-narinaire, embout buccal, facial) qui permet de compenser l’affaiblissement de la fonction respiratoire lorsqu’elle décline.
Qu’est-ce que la maladie de Charcot-Marie-Tooth ?
À ne pas confondre avec la maladie de Charcot que nous venons de décrire ci-dessus, la maladie de Charcot-Marie-Tooth (CMT) est une maladie neurologique héréditaire.
Si elle est plus fréquente que la maladie de Charcot, elle reste cependant moins grave que cette dernière.
Cette maladie génétique se caractérise par une atteinte des nerfs périphériques (qui permettent de relier la moelle épinière aux muscles du corps) des bras et des jambes. Elle démarre généralement au cours de l’enfance ou de l’adolescence mais elle peut également survenir plus tardivement, autour des 50-60 ans.
Il faut savoir que cette maladie touche à peu près 1 personne sur 2 500, soit environ 30 000 personnes en France.
💡 Bon à savoir :
On utilise le terme de maladie de “Charcot-Marie-Tooth” en hommage aux neurologues qui l’ont décrite dans les années 1880, à savoir Jean-Martin Charcot, Pierre Marie et Howard Henry Tooth.
Quels sont les symptômes de la maladie de Charcot-Marie-Tooth ?
Parmi les symptômes les plus fréquents, il est possible de constater :
- une perte de sensibilité dans les pieds et les mains pouvant entraîner une incapacité à ressentir la douleur, la température, les vibrations,… ;
- une déformation des pieds comme des « pieds creux » ou des « orteils en marteau » (qui se recroquevillent vers le bas) ;
- une fonte des muscles au niveau des jambes (mollets, pieds) et au niveau des avant-bras et des mains entraînant ainsi une perte de la force musculaire ;
- des troubles de la marche ;
- des chutes et une certaine maladresse ;
- une paralysie des mains et des jambes.
Chez certaines personnes, il est également possible de constater une atteinte des muscles respiratoires ou des cordes vocales, des troubles visuels et des problèmes de surdité.
Comment se déclenche t-elle ?
Cette maladie est héréditaire, ce qui signifie qu’elle se transmet des parents aux enfants par l’intermédiaire des gènes. En effet, de nombreux gènes à l’origine de la transmission de la maladie ont été détectés au cours de ces dernières années. C’est pour cette raison que les scientifiques disent qu’une cinquantaine de formes de cette maladie existent.
Il est possible classer ces différentes formes selon 3 critères distincts :
- l’anomalie génétique en cause
d’après des recherches, plus d’une soixantaine de gènes peuvent favoriser l’apparition d’une forme de cette maladie ;
- la nature de l’atteinte des nerfs périphériques qui peut se situer soit :
– au niveau des axones des neurones :
un axone correspond à une partie anatomique d’un neurone. Plus précisément, il s’agit de la zone de conduction du message nerveux. Morphologiquement, un axone se caractérise par un long prolongement qui émerge du corps cellulaire du neurone.
– au niveau des gaines de myéline :
les gaines de myéline correspondent à des gaines isolantes qui entourent les axones des neurones afin de les protéger et d’accélérer la conduction des messages nerveux.
- le mode de transmission génétique pouvant relever soit :
– d’une transmission autosomique dominante
le gène d’un seul parent porteur est nécessaire pour que la maladie se développe ;
– d’une transmission autosomique récessive
deux gènes, soit un gène de chaque parent porteur est nécessaire pour que la maladie se développe ;
– d’une transmission liée au chromosome X
le gène anormal à l’origine de la maladie est situé sur le chromosome sexuel X.

